
All materials that are exposed to the outdoor environment are subject to degradation caused by natural weathering processes. Since the mid-19th century, air pollution has been suspected of accelerating the degradation of natural and man-made materials. Limestone and marble are two types of stone used in the construction of buildings and monuments and in the creation of carved stone statuary. Both rock types are largely composed of calcite, the stable polymorph of calcium carbonate (CaCO3).
All carbonate materials are sensitive to acidic deposition. Concern over the effect of pollutants on carbonate stone is directed to the economic consequences of damage to the stone used in construction as well as to the loss of the aesthetic value of historic buildings and monuments. Thus, the erosion of marble or limestone surfaces and the loss of detail on statuary are an important area of concern in research on the effects of acidic deposition. The gradual erosion of carbonate stone is the result of two possibly related processes: carbonate stone dissolution (chemical erosion), and the mechanical loss of grains from stone surfaces (physical erosion).
This paper describes a laboratory experiment on the effects of acidic deposition on carbonate stone erosion. It can serve as the basis for an undergraduate (or precollege) experiment in environmental chemistry. Recent field investigations are described that provide measurements of carbonate stone dissolution and mechanical erosion under weathering conditions that are prevalent in the eastern United States. The purpose of the laboratory work is to answer questions concerning the effects of hydrogen ion deposition on stone erosion processes that were difficult to resolve on the basis of field experiments alone.
Rain water in equilibrium with atmospheric C02 at STP has a pH of 5.6. Natural sources of acidic gases (e.g., volcanic emission Of S02) and organic acids may serve to reduce local rain pH values to between 5.0 and 5.6 (1). In acid rain the primary contributions of hydrogen ion besides the natural sources of acidity are sulfurous, sulfuric, and nitric acids, which lower the pH of rain and accelerate weathering processes. The average pH of most rainfall in the eastern United States is between about 3.9 (Steubenville, OH) and 4.5 (Research Triangle Park, NC), based on rain chemistry data collected by the U.S. Bureau of Mines at field exposure sites for acid rain research (2).
One of the goals of research on the effects of acidic deposition on carbonate stone is to determine the changes in erosion rate in the pH range of 5.0 to 4.0, which reflects the decrease in rain pH due to manmade pollution. In the field of materials effects research, this change in the rate of erosion is called the incremental effect. This term is used throughout the following discussion to signify the amount of additional weathering caused by acidic pollutants, beyond the natural weathering rate expected in pristine environments.
The chemistry of carbonate stone dissolution is treated in numerous texts (3-5), and carbonate stone weathering has been reviewed in several books (6-8) and review papers (9-10). Under equilibrium conditions, the incremental impact of hydrogen ion in pH 4.0 rainwater on carbonate stone dissolution is expected to be small because the equilibrium solubility of calcite is dominated by its reaction with carbonic acid derived from atmospheric C02 that has dissolved in rainwater.
The effect of solution acidity on the dissolution of carbonate stone can be predicted using an equilibrium computer model (12). The equilibrium calcium concentrations calculated are
0.582 mmol/L for rainwater at pH 4.00.551 mmol/L for rainwater at pH 5.0
Thus, for pH 4.0 rain, the predicted incremental effect of the deposited hydrogen ion would be an approximate 6% increase in calcite dissolution (100 x (0.582-0551)/0.551).
However, solutions collected from run-off experiments at field exposure sites for acid rain materials research (13), and from in-situ experiments conducted on carbonate stone monuments, are not saturated in calcium carbonate. The dissolution of carbonate stone in undersaturated systems has been studied extensively (14).
As water flows across a stone surface, the usual model of the interaction process assumes that the exterior surface of the stone is separated from the bulk solution by a thin fluid boundary layer (15). Numerous laboratory experiments to investigate the kinetics of calcite dissolution have shown that under nonequilibrium conditions, the dissolution rate of carbonate stone is dependent not only on the kinetics of the reactions at the stone surface but also on the transport of hydrogen ion, bicarbonate ion, calcium ion, and carbon dioxide gas across the boundary layer. In addition, carbon dioxide gas transfer at the solution/air surface influences the net dissolution reaction.
If carbonate stone dissolution is controlled by reaction kinetics, the incremental impact of hydrogen ion could be quite significant because the dissolution rate would be controlled by the rate of transport of hydrogen ion across the fluid boundary layer. Calculations from a rate equation for calcite dissolution (14) predict an approximate 8-fold difference in the initial calcite dissolution rate over the pH range 5.0 to 4.0.
Due to the potential importance of reaction kinetics and the dynamics of fluid flow across the stone surface (particularly for low pH solutions at high flow rates), the incremental impact of hydrogen ion on carbonate stone dissolution is difficult to predict. Thus, direct experimental measurements are required to define the importance of kinetic and hydrodynamic factors.
The effects of acidic deposition on carbonate stone have been the subject of laboratory and field experiments. The effects of acid rain on stone monuments have been reviewed previously in this Journal by Charola (16). A thorough review of stone weathering processes and of the research on the effects of pollutants on stone surfaces is provided by Amoroso and Fassina (17). Lipfert (18) has reviewed a previously published work that reports quantitative measurements of stone weathering rates.

Figure 1. Scatter plot of "excess" calcium concentration (blank corrected) in marble run-off from the New York exposure site against hydrogen ion concentration in the incident rain as measured by a run-off blank. The calcium concentrations have been corrected for dry deposition effects and temperature. The lines represent reaction of the stone with hydrogen ion to produce bicarbonate (dashed line) and CO2(gas) (solid line).
Most of the previous work on the effects of acid rain on stone erosion has relied on measurements of damage to test samples (such as weight loss measurements), coupled with concomitant measurements of environmental conditions during the period of exposure. Statistical analysis has then been used to derive the relationships between rates of deposition and erosion. The emphasis of current research on acidic deposition (such as the experiments described here) is in making quantitative measurements of stone damage processes rather than relying on statistical inference of cause and effect.
During the past several years, research in the effects of acidic deposition on carbonate stone has been conducted under the National Acid Precipitation Assessment Program (NAPAP) (2) to define the incremental effect of acidic deposition under ambient environmental conditions. In experiments conducted at field exposure sites, the concentration of calcium ions in run-off solutions from marble and limestone slabs (1 ft. x 2 ft. x 2 in.) was used to measure the amount of stone erosion due to dissolution (19).
The most direct approach to demonstrating the effect of the deposition of hydrogen ion on carbonate stone surfaces is to prepare a plot of calcium ion concentration (corrected for the effects of dry deposition) in the run-off solutions against hydrogen ion concentration in the incident rain. A typical plot is shown in Figure 1. The nonzero intercept reflects the solubility of calcium carbonate in "clean rain", that is, the solubility of calcium carbonate in water that is in equilibrium with atmospheric C02. The approximate intercept in Figure 1 of 0.13 mmol/L is at least 4 times lower than the predicted solubility of of 0.55 mmol/L, showing that the run-off solutions are undersaturated.
The incremental effect of hydrogen ion deposition on the erosion of carbonate stone surfaces can be defined by the measured hydrogen ion concentration of the rain and the stoichiometry of the reaction of H+ with CaCO3. In Figure 1 two lines define the stoichiometry of two possible reactions of CaCO3 with H+. In run-off solutions from carbonate stone surfaces the principal carbonate species is bicarbonate ion, and the line of highest slope (dashed line in Fig. 1) represents the reaction that yields HCO3-.
Thus, the predicted effect of the wet deposition of hydrogen ion on carbonate stone would be 1 mol of calcite dissolved for each mole of hydrogen ion deposited.
The line of lower slope (solid line in Fig. 1) represents the reaction when C02(gas) is lost from the system:
This reaction reduces the effect of hydrogen ion deposition because 1 mol of calcite would be dissolved for every 2 mol of hydrogen ion deposited. Thus, the predicted stoichiometry of the reaction of carbonate stone with hydrogen ion would be between 1 and 2 depending on the degree of outgassing.
Considerable scatter makes it difficult to define the effect of hydrogen ion between the two limiting cases. Much of the scatter may be caused by changes in flow rate across the stone surfaces during the course of a rain event. Variations in rain pH during a rain event may also contribute to the scatter because the earliest rain tends to be the most acidic.
As noted above, the run-off experiments measure only stone loss through dissolution. However, the mechanical removal of granular material from the stone surface may also contribute to the stone erosion process. In addition to the run-off experiments, the NAPAP experiments included two additional measurements of stone erosion:
Changes in the weight of stone test briquettes (3 in. x 3.5 in. x 2 in.) were used to measure total erosion caused by dissolution and physical grain loss (20).
A spectroscopic method, holographic laser interferometry, was used to obtain an erosion profile of test samples, providing an independent measure of the total erosion process (21).
The results from the weight loss and interferometry experiments showed that the rate of erosion of stone exceeded the rate of stone loss due to dissolution by nearly a factor of 2 for marble and nearly 3 for limestone (19). The large magnitude of the grain loss process relative to the effects of dissolution was an unexpected result from the field experiments. Thus, it is important to attempt to quantify any possible relationship between acidic deposition and the loss of granular material.
A series of laboratory run-off experiments were undertaken to help resolve difficult questions using the field data.
Is the stoichiometry of the hydrogen ion reaction described by eq 3 or by eq 4, or is the effect intermediate between these two limiting cases?Is the rate of grain loss controlled, at least in part, by the pH of the incident rain?
In addition, we were interested in making a qualitative estimate of the effect of droplet residence time (the time that a drop of water is in contact with the stone surface) on stone dissolution.
Test slabs of Shelburne marble from Vermont (used in the Jefferson Memorial, Washington, DC) and salem limestone from Indiana (used in the National Cathedral, Washington, DC) were used for the experiments reported here. The stone slabs were identical to those used at the NAPAP field exposure sites.
The slabs were selected because they are similar in appearance and physical characteristics to the stone used in many monuments and government and private buildings in the eastern and Midwestern United States. The slabs used measured 1 ft. x 2 ft. x 2 in., although satisfactory results can be obtained using smaller slabs, as shown below. (Suitable pieces of marble and limestone can be obtained from funeral monument stone companies or dealers of decorative and construction stone.)
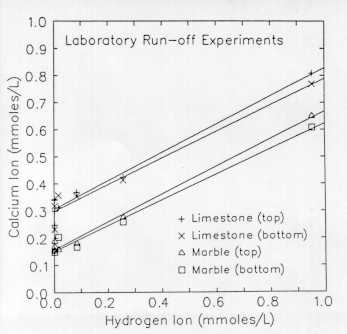
Figure 2. Scafter plot of calcium concentration in marble and limestone run-off from laboratory experiments against hydrogen ion concentration in the incident spray. The spray solutions were deionized water acidified with sulfuric acid. The lines are least-squares regression lines through the data. The spray was applied to either the top or bottom half of the inclined stone slabs in separate experiments to test for possible effects of droplet residence time.
The slabs were held in a wood frame at an inclination of 30' to horizontal. A series of four solutions, ranging in pH from 4.8 to 3.0, were prepared in increments of approximately 0.5 pH unit with deionized water acidified with sulfuric acid. Then 500 mL of each solution was sprayed evenly over the stone slabs with a small garden sprayer, and the run-off solutions were collected in 1-ft.-long plastic troughs mounted at the base of each slab.
Two sets of experiments were run to provide a qualitative test of droplet residence time. In the first set, only the upper 1-ft. x 1-ft. area of the 1-ft. x 2-ft. test slab was sprayed, and the collected solution ran over most of the 2-ft2 surface. In the second set, the spray was restricted to the bottom 1-ft. x 1-ft. area.
The slabs were first sprayed with distilled water with a pH of 5.6, and then the acidified solutions were sprayed in order of decreasing pH. Then the slabs were again sprayed with distilled water. The slabs were allowed to dry for at least 1 day between spraying with each solution.
Each run-off solution was filtered through a Nucleopore filter. The filters were dried and weighed in order to measure the amounts of granular material removed from the stone during each spraying of the stone slabs. The volume of the run-off was measured, and the calcium concentration in the run-off solution was determined by atomic absorption. Calibration standards, ranging in calcium concentration from 5 to 40 mg/L, were prepared by diluting a 500-mg/L stock solution prepared by dissolving calcium carbonate as a primary standard.
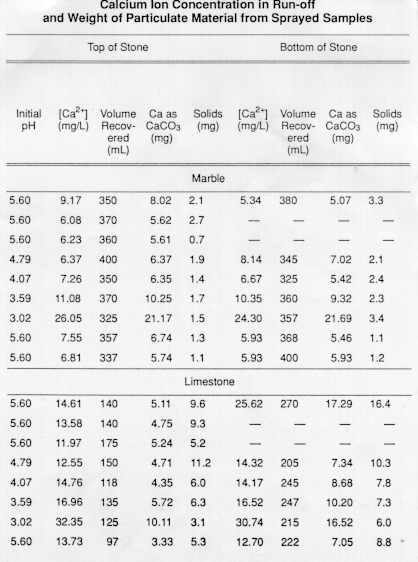
The results show that the concentration of calcium in the run-off solutions is largest for incident spray having a low pH (see the table). The calcium concentration varies linearly with initial hydrogen ion concentration of the spray solutions (Fig. 2). The slope of the regression lines is approximately 0.5, as predicted by the 2:1 stoichiometry (2H+:1CaCO3)of eq 4.
The slightly lower slope for the samples for which only the bottom half of the slab was sprayed demonstrates that a small percentage of the hydrogen ion in the incident spray falling near the bottom edge of the stone slab was not neutralized by reaction with the stone.
The larger intercept values for limestone, as compared to marble, show that the water in contact with limestone more closely approaches the equilibrium concentration of 0.55 mmol/L due to limestone's greater porosity and larger surface area.
The data from Figure 2 can be used to define simple quantitative relationships for the effect of hydrogen ion deposition on the chemical erosion of carbonate stone surfaces. The slope of the regression line represents the effect of hydrogen ion, and the intercepts are measures of the clean rain" solubility of limestone and marble.
Thus, for marble we get
and for limestone,
where the concentration values are in mmol/L.
For the average rain pH of 4.2 at the NAPAP exposure sites (except for Steubenville, Ohio), hydrogen ion deposition would contribute 17% to the chemical erosion of marble and 10% to limestone weathering for the flow rate conditions defined by the experimental design. Under different flow rate conditions, of course, the incremental effect of hydrogen ion deposition could vary due to the kinetics of carbonate dissolution.
In each experimental run, 500 mL of solution was sprayed on the stone, but smaller volumes were recovered. (See the columns 'Volume Recovered" in the table.) The average volumes collected were:
The large differences in run-off volume between the marble and limestone reflect the magnitude of the differences in water retention by the two types of stone. This is due to the much greater porosity of the limestone. In spite of the higher concentrations of Ca2+, in the limestone runoff, the total calcium removed from the stone by the dissolution process is smaller for limestone than for marble, because more of the incident water is retained by the limestone slab. (See "Ca as CaCO3+ in the table.) The smaller run-off volumes for limestone when the top half of the slab was sprayed are due to the longer flow path and larger surface area exposed to the solutions.
The total amount of stone lost through dissolution (listed under "Ca as CaCO3" in the table) and the weight of granular material collected on the Nucleopore filters (listed under "Solids" in the table) may be compared with the field observations reported in the previous section. For limestone slabs, the amount of granular material recovered decreased during the course of the experiments. This is possibly due to the initial presence of loosely held grains on the surface that were gradually washed away during the experiments.
The average amount of grain loss in the last few runs was roughly equivalent to the amount of calcium removed by dissolution at a typical rain pH of about 4.0 (i.e., "Ca as CaCO3" for pH 4.07 in the table). This observation is broadly consistent with the field data. In contrast, the average amount of granular material collected from the marble slabs was approximately 4 times lower than the amount of stone lost by dissolution at pH 4.07. The NAPAP field data do not support as large a difference in grain loss between limestone and marble as the laboratory results. The field data, however, represent the results of stone exposed to the full range of ambient weathering conditions:
large temperature fluctuationsvariations in rainfall intensity
the effects of dry deposition
A primary goal of the laboratory experiment was to test for a possible relationship between the amount of granular material lost from the stone and the acidity of the incident spray. If grain loss is due to selective dissolution of the carbonate matrix, then grain removal should be proportional to chemical erosion, and thus an amplification of it. Then grain loss can be partially attributed to the effects of acidic deposition. Conversely, if grain loss can be ascribed to mechanical causes (impact of rainfall, water flow, freezethaw cycles), then this process may be independent of pollutant deposition. A significant finding of the laboratory experiments is an absence of a pH dependence for the amount of granular material collected from either the marble or limestone slabs.
Under ambient conditions, grain loss may also be influenced by dry acidic deposition, such as the deposition of sulfur dioxide between rainfall events to form gypsum. The possible importance of dry deposition to the grain removal process remains an open question in the weathering of carbonate stone (19). The reprecipitation of calcite from solutions trapped in the stone pores, particularly in porous limestone surfaces, also increases the complexity of the grain loss process.
One of the goals of research on the effects of acidic deposition on carbonate stone surfaces is to define the incremental impact of acidic deposition relative to natural weathering processes on the rate of carbonate stone erosion. If rain that impacts carbonate stone surfaces is resident on the surface long enough to approach chemical equilibrium, the incremental effect of hydrogen ion is expected to be small (i.e., 6% for a rain of pH 4.0). Under nonequilibrium (i.e., high flow rate) conditions, kinetic considerations suggest that the incremental effect of hydrogen ion deposition could be quite significant.
Field run-off experiments involving the chemical analysis of rain collected from inclined stone slabs have been used to evaluate stone dissolution processes under ambient conditions of wet and dry deposition of acidic species.
The stoichiometry of the reaction of stone with hydrogen ion is difficult to define from the field data due to scatter in the data attributed to hydrodynamic effects.
Laboratory run-off experiments show that the stoichiometry is best defined by a reaction with H+ in which C02 is released from the system. The baseline effect caused by water in equilibrium with atmospheric C02 is identical in the field and in laboratory simulation. The experiments show that the solutions are close enough to equilibrium for the incremental effect of hydrogen ion to be minor (i.e., 24% for marble for a rain of pH 4.0) relative to dissolution due to water and carbonic acid reactions.
Stone erosion rates based on physical measurement are approximately double the recession rates that are due to dissolution (estimated from the observed calcium content of the run-off solutions). The difference may reflect the loss of granular material not included in recession estimates based on the run-off data. Neither the field nor the laboratory run-off experiments indicate a pH dependence for the grain-removal process.
P. Baedecker wishes to acknowledge the assistance of Cheryl E. Baedecker and Mary Jo Baedecker during the initial development of the laboratory experiments. The authors also wish to thank P. D. Glynn, J. W. Morgan, V. G. Mossotti, and two anonymous persons for helpful reviews.
1. Charlson, R. J.; Rodhe, H. Nature 1982, 295, 683-685.2. Baedecker, P. A.; Edney, E. O.; Moran, P. J.; Simpson, T. C.; Williams, R. S.; Hoaker, R. P; Kishiyama, G.; Langmuir, D.; McGee, E. S.; Mossotti, V G.; Pavich, M. J.; Reddy, M. M.; Reimann, K J.; Schmiernund, R.; Sciammarella, C. A.; Spiker, E. C.; Weseley, M. L.; Youngdahl, C. A. Effects of acidic Deposition on Materials; NAPAP Report 19, Acidic Deposition: State of Science and Technology; National Acid Precipitation Assemment Program, 722 Jackson Place, NW, Washington, DC, 1990.
3. Garrels, R. M.; Christ, C. L. Solutions, Minerals, and Equilibria; Harper and Row: New York, 1965; p 450.
4. Krauskopf, K. B. Introduction to Geochemistry; McGraw-Hill. New York, 1967; p 721.
5. Stumm, W.; Morgan, J. J. Aquatic Chemistry, Znd ad.; John Wiley: New York, 1981; p 780.
6. Winkler, E. M. Stone: Properties, Durability in Man's Environment, 2nd ed.; Springer-Verlag: New York, 1975; p 230.
7. Jennings, J. N. Karst Geomorphology; Basil Blackwell: Oxford, 1985; p 293.
8. White, W. B. Geomophology and Hydrology of Karst Terrains; Oxford University: New York, 1988; p 464.
9. Keller, W. C. In Decay and Preservation of Stone; Winkler, E. M.: Ed.; Engineering Geology Case Histories, Number 11, Geological Soc. Am, Boulder, CO, 1978: pp 37-46.
10. Jennings, J. N. Am. Scientist 1983, 71, 578-586.
11. Kem, E. M. J. Chem. Edw. 1960, 37,14-22.
12. Parkhurst, D. L.; Thoratenson, D. C.; Plummer, L N. PHREEQE-A computer program for geochemical calculations; U.S. Geol. Survey Water Res. Invest., 1990; p 80-96.
13. Reddy, M. M. Earth Surface Processes and Landforms 1988, 13, 335-354.
14. Plummer, L. N.; Parkhurst, D. L.; Wigley, T M. L. In Chemical modelling of aqueous systems; Jenne, E. A.: Ed.; Am. Chem. Soc. Symp. Ser. 93., Am. Chem. Soc.: Washington, DC: 1979; pp 537-673.
15. Cussler, E. L. Diffusion, Mass Transfer in Fluid Systems; Cambridge Univ.: New York, NY, 1984; p 525.
16. Charola, A. E., J. Chem. Educ. 1987, 64, 436-437.
17. Amoroso, G.; Fassina, V. Stone decay and conversation; Materials Sci. Monographs, 11, Elsevier. New York, 1983; p 453.
18. Lipfert, F. W. Atmos. Environ. 1989, 23, 415-429.
19. Baedecker, P. A.; Reddy, M. M.; Raimann, K. J.; Sciammarelia, C. A. Atmos. Environ., 1992; 26B, 147-158.
20. Youngdahl, C. A.; Doe, B. R. In Materials Degradation Caused by Acid Rain; Baboian, K: Ed.; ACS Symp. Series, Vol. 318., Am. Chem. Soc. Washington, DC: 1986; pp 226-238.
21. Sciammarella, C. Opt. Eng. 1982, 21, 447-457.
This article was published in the Journal of Chemical Education, February 1993, Volume 70, Number 2. pp. 104-108.