| Next || Previous || Top |
Description of Data Input
The input for PHREEQC is arranged by keyword data blocks. Each data block begins with a line that contains the keyword (and possibly additional data) followed by additional lines containing data related to the keyword. The keywords that define the input data for running the program are listed in table 1. Keywords and their associated data are read from a database file at the beginning of a run to define the elements, exchange reactions, surface complexation reactions, mineral phases, gas components, and rate expressions. Any data items read from the database file can be redefined by keyword data blocks in the input file. After the database file is read, data are read from the input file until the first END keyword is encountered, after which the specified calculations are performed. The process of reading data from the input file until an END, followed by doing the calculations, is repeated until the end of the input file is encountered. The set of calculations, defined by keyword data blocks terminated by an END, is termed a “simulation”. A “run” is a series of one or more simulations that are contained in the same input data file and calculated during the same invocation of the program PHREEQC.
Table 1. List of keyword data blocks.
|
Keyword data block
|
Function
|
|
ADVECTION
|
Specify parameters for advective-reactive transport, no dispersion
|
|
CALCULATE_VALUES
|
Define Basic functions
|
|
COPY
|
Make copies of keyword data blocks with new identifying numbers
|
|
DATABASE
|
Specify the database for the simulations
|
|
DELETE
|
Delete specified reactants
|
|
DUMP
|
Write complete descriptions of specified reactants to file (or to a string for IPhreeqc modules)
|
|
END
|
Demarcate end of a simulation
|
|
EQUILIBRIUM_PHASES
|
Define assemblage of minerals and gases to react with an aqueous solution
|
|
EXCHANGE
|
Define exchange assemblage composition
|
|
EXCHANGE_MASTER_SPECIES
|
Identify exchange sites and corresponding exchange master species
|
|
EXCHANGE_SPECIES
|
Define association half-reaction and thermodynamic data for exchange species
|
|
GAS_PHASE
|
Define a gas-phase composition
|
|
INCLUDE$
|
Insert a file into the input or database file
|
|
INCREMENTAL_REACTIONS
|
Define whether reaction increments are incremental or cumulative
|
|
INVERSE_MODELING
|
Specify solutions, reactants, and parameters for mole-balance modeling
|
|
ISOTOPES
|
Identify isotopes of elements and define the absolute isotopic ratio of standards
|
|
ISOTOPE_ALPHAS
|
Specify fractionation factors to appear in the output file
|
|
ISOTOPE_RATIOS
|
Specify isotope ratios to appear in the output file
|
|
KINETICS
|
Specify kinetic reactions and define parameters
|
|
KNOBS
|
Define parameters for numerical method and printing debugging information
|
|
LLNL_AQUEOUS_MODEL_PARAMETERS
|
Specify activity coefficient parameters for the Lawrence Livermore National Laboratory aqueous model
|
|
MIX
|
Define mixing fractions of aqueous solutions
|
|
NAMED_EXPRESSIONS
|
Assigns a name to an analytical expression for an equilibrium constant or isotope fractionation factor
|
|
PHASES
|
Define dissociation reactions and thermodynamic data for minerals and gases
|
|
PITZER
|
Specify the parameters of a Pitzer specific-ion-interaction aqueous model
|
|
PRINT
|
Select data blocks to be printed to the output file
|
|
RATES
|
Define rate equations with Basic language statements
|
|
REACTION
|
Specify irreversible reactions
|
|
REACTION_PRESSURE
|
Specify pressure(s) for batch reactions
|
|
REACTION_TEMPERATURE
|
Specify temperature(s) for batch reactions
|
|
RUN_CELLS
|
Specify a reaction simulation that includes all reactants of a given identification number
|
|
SAVE
|
Save results of batch reactions for use in subsequent simulations
|
|
SELECTED_OUTPUT
|
Print specified quantities to a user-defined file
|
|
SIT
|
Specify the parameters of a SIT (Specific ion Interaction Theory) aqueous model
|
|
SOLID_SOLUTIONS
|
Define the composition of a solid-solution assemblage
|
|
SOLUTION
|
Define the composition of an aqueous solution
|
|
SOLUTION_MASTER_SPECIES
|
Identify elements and corresponding aqueous master species
|
|
SOLUTION_SPECIES
|
Define association reaction and thermodynamic data for aqueous species
|
|
SOLUTION_SPREAD
|
Define one or more aqueous solution compositions using a tab-delimited format (Alternative input format for
SOLUTION
)
|
|
SURFACE
|
Define the composition of an assemblage of surfaces
|
|
SURFACE_MASTER_SPECIES
|
Identify surface sites and corresponding surface master species
|
|
SURFACE_SPECIES
|
Define association reaction and thermodynamic data for surface species
|
|
TITLE
|
Specify a text string to be printed in the output file
|
|
TRANSPORT
|
Specify parameters for advective-dispersive-reactive transport, optionally with dual porosity
|
|
USE
|
Select aqueous solution or other reactants that define batch reactions
|
|
USER_GRAPH
|
Specify data and parameters for a user-defined X-Y plot
|
|
USER_PRINT
|
Print user-defined quantities to the output file
|
|
USER_PUNCH
|
Print user-defined quantities to the selected-output file
|
Each simulation may contain one or more of seven types of speciation, batch-reaction, and transport calculations: (1) initial solution speciation, (2) determination of the composition of an exchange assemblage in equilibrium with a fixed solution composition, (3) determination of the composition of a surface assemblage in equilibrium with a fixed solution composition, (4) determination of the composition of a fixed-volume gas phase in equilibrium with a fixed solution composition, (5) calculation of chemical composition as a result of batch reactions, which include mixing; kinetically controlled reactions; net addition or removal of elements from solution, termed “net stoichiometric reaction”; variation in temperature and pressure; equilibration with assemblages of pure phases, exchangers, surfaces, and (or) solid solutions; and equilibration with a gas phase at a fixed total pressure or fixed volume, (6) advective-reactive transport, or (7) advective-dispersive-reactive transport. The combination of capabilities allows the modeling of complex geochemical reactions and transport processes during one or more simulations.
In addition to speciation, batch-reaction, and transport calculations, the code may be used for inverse modeling, by which net chemical reactions are deduced that account for composition differences between an initial water or a mixture of initial waters and a final water.
Conventions for Data Input
PHREEQC was designed to eliminate some of the input errors due to complicated data formatting in Fortran-type input files. Data for the program are free format; spaces or tabs may be used to delimit input fields (except SOLUTION_SPREAD, which is delimited only with tabs); blank lines are ignored. Keyword data blocks within a simulation may be entered in any order. However, data elements entered on a single line are order specific. As much as possible, the program is case insensitive. However, chemical formulas are case sensitive.
The following conventions are used for data input to PHREEQC:
Keywords
--Input data blocks are identified with an initial keyword. This word must be spelled exactly, although case is not important. Several of the keywords have synonyms. For example,
PURE_PHASES
is a synonym for EQUILIBRIUM_PHASES.
Identifiers
--Identifiers are options that may be used within a keyword data block. Identifiers may have two forms: (1) they may be spelled completely and exactly (case insensitive) or (2) they may be preceded by a hyphen and then only enough characters to uniquely define the identifier are needed. The form with the hyphen is always acceptable and is recommended. Usually, the form without the hyphen is acceptable, but in some cases the hyphen is needed to indicate the word is an identifier rather than an identically spelled keyword; these cases are noted in the definitions of the identifiers in the following sections. In this report, the form with the hyphen is used except for identifiers of the SOLUTION keyword and the identifiers
log_k
and
delta_h
. The hyphen in the identifier never implies that the negative of a quantity is entered.
Chemical equations
--For aqueous, exchange, and surface species, chemical reactions must be
association
reactions, with the defined species occurring in the first position after the equal sign. For phases, chemical reactions must be
dissolution
reactions with the formula for the defined phase occurring in the first position on the left-hand side of the equation. Additional terms on the left-hand side are allowed. All chemical equations must contain an equal sign, “=”. In addition, left- and right-hand sides of all chemical equations must balance in numbers of atoms of each element and total charge. All equations are checked for these criteria at runtime, unless they are specifically excepted. Nested parentheses in chemical formulas are acceptable. Spaces and tabs within chemical equations are ignored. Waters of hydration and other chemical formulas (that are normally represented by a “ · ”, as in the formula for gypsum, CaSO
4
·2H
2
O) are designated with a colon (“:”) in PHREEQC (thus, CaSO
4
:2H
2
O), but only one colon per formula is permitted.
Element names
--Two forms of element names are available (1) those beginning with an alphabetic character and (2) those beginning with a square bracket. For form 1, an element formula, wherever it is used, must begin with a capital letter and may be followed by one or more lowercase letters or underscores, “_”. Numbers are not permitted, except in parentheses for defining the redox state. In general, element names are simply the chemical symbols for elements, which have a capital letter and zero or one lower case letter. It is sometimes useful to define other entities as elements, which allows mole balance and mass-action equations to be applied. Thus, “Fulvate” is an acceptable element name, and it would be possible to define metal binding constants in terms of metal-Fulvate complexes.
Form 2 of element names is less restrictive than form 1. Within the square brackets, any combination of alphanumeric characters and the characters plus, minus, equal, colon, decimal point, and underscore can be used. The form-2 element name is case dependent, but upper and lower case characters can be used in any position. The iso.dat database makes extensive use of the square-bracket form for element names by using the mass number and chemical symbol for minor-isotope definitions, such as [13C], [15N], and [34S].
Charge on a chemical species
--The charge on a species may be defined by the proper number of pluses or minuses following the chemical formula or by a single plus or minus followed by an integer designating the charge. Either of the following is acceptable, Al+3 or Al+++. However, Al3+ would be interpreted as a molecule with three aluminum atoms and a charge of plus one.
Valence states
--Redox elements that exist in more than one valence state in solution are identified for definition of solution composition by the element name followed by a valence in parentheses. Thus, sulfur that exists as sulfate is defined as S(6) and total sulfide (H
2
S, HS
-
, and others) is identified by S(-2). The valence may include a decimal point. The valence number is for identification purposes only and does not otherwise affect the calculations.
log K and temperature dependence
--The identifier
log_k
is used to define the log
K
at 25 °C for a reaction. The temperature dependence for log
K
may be defined by the Van’t Hoff expression or by an analytical expression. The identifier
delta_h
is used to give the standard enthalpy of reaction at 25 °C for a chemical reaction, which is used in the Van’t Hoff equation. By default the units of the standard enthalpy are kilojoule per mole (kJ/mol). Optionally, for each reaction the units may be defined to be kilocalorie per mole (kcal/mol). An analytical expression for the temperature dependence of log
K
for a reaction may be defined with the
-analytical_expression
identifier. Up to six numbers may be given, which are the coefficients for the equation: 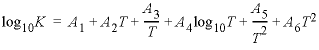 , where T is in kelvin. A log
K
is defined either with
log_k
or
-analytical_expression
(default
log_k
is zero); the enthalpy is optional (default is zero). If present, an analytical expression is used in preference to the
log_k
and enthalpy values for calculation of the log
K
at the specified temperature.
, where T is in kelvin. A log
K
is defined either with
log_k
or
-analytical_expression
(default
log_k
is zero); the enthalpy is optional (default is zero). If present, an analytical expression is used in preference to the
log_k
and enthalpy values for calculation of the log
K
at the specified temperature.
Pressure dependence of log K
--Pressure dependency of reaction constants for species, and the pressure-dependent solubilities of minerals and gases, are calculated from the volume change of the reaction. The molar volume of solids and parameters for calculating the molal volume of aqueous species are defined in Amm.dat, phreeqc.dat, and pitzer.dat.
Comments
--The “#” character delimits the beginning of a comment in the input file. All characters in the line that follow this character are ignored. If the entire line is a comment, the line is not echoed to the output file. If the comment follows input data on a line, the entire line, including the comment, is echoed to the output file. The “#” is useful for adding comments explaining the source of various data or describing the problem setup. In addition, it is useful for temporarily removing lines from an input file.
Logical line separator
--A semicolon (“;”) is interpreted as a logical end-of-line character. This allows multiple logical lines to be entered on the same physical line. For example, solution data could be entered as:
pH 7.0; pe 4.0; temp 25.0
on one line. The semicolon should not be used in character fields, such as the title or other comment or description fields.
Logical line continuation
--A backslash (“\”) at the end of a line may be used to merge two physical lines into one logical line. For example, a long chemical equation could be entered as:
Ca0.165Al2.33Si3.67O10(OH)2 + 12 H2O = \
0.165Ca+2 + 2.33 Al(OH)4- + 3.67 H4SiO4 + 2 H+
on two lines. The program would interpret this sequence as a balanced equation entered on a single logical line. For a line to be logically continued, the backslash must be the last character in the line except for white space.
Repeat count
--An asterisk (“*”) can be used to indicate a repeat count for the data item that follows the asterisk. The format is an integer followed directly by the asterisk, which is followed directly by a numeric value. For example “4*1.0” is the same as entering four values of 1.0 (“1.0 1.0 1.0 1.0”). Repeat counts can be used for specifying data for the identifiers
-length
and
-dispersivity
in the TRANSPORT data block and for specifying reaction steps in the REACTION and KINETICS data blocks.
Range of integers
--A hyphen (“-”) can be used to indicate a range of integers for the keywords with an identification number (for example, SOLUTION 2-5). It is also possible to define a range of cell numbers for the identifiers
-print_cells
and
-punch_cells
in the ADVECTION and TRANSPORT data blocks and in the options for the COPY, DELETE, DUMP, and RUN_CELLS data blocks. A range of integers is given in the form
m-n
, where
m
and
n
are positive integers,
m
is less than
n
, and the two numbers are separated by a hyphen without intervening spaces.
Special characters
--A summary of all of the special characters used in PHREEQC formatting is given in table 2.
Table 2. Summary of special characters for input data files.
|
Special character
|
Use
|
-
|
When preceding a character string, a hyphen indicates an identifier (option) for a keyword.
|
-
|
Indicates a range of cell numbers for keyword data blocks (for example,
SOLUTION
2-5), for identifiers
-print_cells
and
-punch_cells
in the
ADVECTION
and
TRANSPORT
data blocks, and for identifiers in the
COPY
,
DELETE
,
DUMP
, and
RUN_CELLS
data blocks.
|
:
|
In a chemical equation, “:” replaces “
.
” in a formula like CaSO
4
.
2H
2
O.
|
[ ]
|
Used to define element names including numeric and a limited set of special characters (+-._:).
|
( )
|
The redox state of an element is defined by a valence enclosed by parentheses following an element name.
|
#
|
Comment character, all characters following # are ignored.
|
;
|
Logical line separator.
|
\
|
Line continuation if “\” is the last non-white-space character of a line.
|
*
|
Can be used to indicate a repeat count for
-length
and
-dispersivity
values in the
TRANSPORT
data block and steps in the
REACTION
and
KINETICS
blocks.
|
Reducing Chemical Equations to a Standard Form
The numerical algorithm of PHREEQC requires that chemical equations be written in a particular form. Internally, every equation must be written in terms of a minimum set of chemical species; essentially, one species for each element or valence state of an element. For the program PHREEQE, these species were called “master species” and the reactions for all aqueous complexes had to be written using only these species. PHREEQC also needs reactions in terms of master species; however, the program contains the logic to rewrite the input equations into this form. Thus, it is possible to enter an association reaction and log K for an aqueous species in terms of any aqueous species in the database (not just master species), and PHREEQC will rewrite the equation to the proper internal form.
PHREEQC also will rewrite reactions for phases, exchange complexes, and surface complexes. Reactions are required to be dissolution reactions for phases and association reactions for aqueous, exchange, or surface complexes. Dissolution reactions for phases allow inclusion of names of solids and gases in the equations, provided they are appended with the strings “(s)” and “(g)”; for example,
CaCO2[18O](s) + H2O(l) = H2[18O](aq) + Calcite(s).
The string “(l)” can be appended to the water formula and “(aq)” to aqueous species for clarity, but they are not required. The “(s)” and “(g)” suffixes cause the program to look in the list of phases to find equations that can be used to reduce the original equation to an equation that contains exclusively aqueous species. This capability to use solids and gases in chemical reactions for phases was implemented primarily to simplify the definition of equations for isotopic solid and gas components. The log Ks for these isotopic species often depend on the log K for the predominant isotopic species (solid or gas) offset by a fractionation factor and (or) a symmetry-derived log K. The inclusion of gases and solids in the equations for isotopic solids and gases is a straightforward method to define these dependencies of the isotopic species equilibrium constant on the equilibrium constant for the predominant isotopic species. In the example given here, the equilibrium constant for the single oxygen-18 form of calcium carbonate solid depends on the equilibrium constant of the pure carbon-12, oxygen-16 form of calcite, which is specified by “Calcite(s)” in the example equation and refers to the equation and log K defined for the calcite phase.
There is one major restriction on the rewriting capabilities for aqueous species. PHREEQC calculates mole balances on individual valence states or combinations of valence states of an element for initial solution calculations. It is necessary for PHREEQC to be able to determine the valence state of an element in a species from the chemical equation that defines the species. To do this, the program requires that only one aqueous species of an element valence state is defined by the electron half-reaction that relates it to another valence state. The aqueous species defined by this half-reaction is termed a “secondary master species”; there must be a one-to-one correspondence between valence states and secondary master species and the coefficient of the newly defined species must be one. In addition, there must be one “primary master species” for each element, such that reactions for all aqueous species for an element can be rewritten in terms of the primary master species. The equation for the primary master species is simply an identity reaction. If the element is a redox element, the primary master species must also be a secondary master species. For example, to be able to calculate mole balances on total iron, total ferric iron, or total ferrous iron, a primary master species must be defined for Fe (iron) and secondary master species must be defined for Fe(+3) (ferric iron) and Fe(+2) (ferrous iron). In the default databases, the primary master species for Fe is Fe
+2
, the secondary master species for Fe(+2) is Fe
+2
, and the secondary master species for Fe(+3) is Fe
+3
. The correspondence between master species and elements and element valence states is defined by the
SOLUTION_MASTER_SPECIES
data block, which for iron in phreeqc.dat is as follows:
SOLUTION_MASTER_SPECIES
Fe Fe+2 0.0 Fe 55.847
Fe(+2) Fe+2 0.0 Fe
Fe(+3) Fe+3 -2.0 Fe
The line with “Fe” (without parentheses) defines the primary master species, and the last two lines, which have parentheses following “Fe”, define the secondary master species. The chemical equations for the master species and all other aqueous species are defined by the
SOLUTION_SPECIES
data block.
Conventions for Documentation
The descriptions of keywords and their associated input are now described in alphabetical order as listed in table 1. Several formatting conventions are used to help the user interpret the input requirements. In this report, keywords are always capitalized and bold. Words in bold must be included literally when creating input files (although upper and lower case are interchangeable and optional spellings may be permitted). “Identifiers” are additional keywords that apply only within a given keyword data block; they can be considered to be sub-keywords or options. Although identifiers are case independent, lowercase bold is used in this report for all identifiers except
pH
,
-Donnan
,
-multi_D
, and
-interlayer_D
, for which mixed case is used. “
temperature
” is an identifier for
SOLUTION
input. Each identifier may have two forms: (1) the identifier word spelled exactly (“
temperature
”, in this case), or (2) a hyphen followed by a sufficient number of characters to define the identifier uniquely (for example,
-t
for temperature in
SOLUTION
the data block.). The form with the hyphen is recommended. Words in
italics
are input values that are variable and depend on user selection of appropriate values. Items in brackets ([ ]) are optional input fields. Mutually exclusive input fields are enclosed in parentheses and separated by the word “or”. In general, the optional fields in a line must be entered in the specified order, but it is sometimes possible to omit intervening fields. For clarity, commas sometimes are used to delimit input fields in the explanations of data input; however, commas are not allowed in the input data file except in Basic programs; in all other cases, only white space (spaces and tabs) may be used to delimit fields in input files. Where applicable, default values for input fields are stated.
Getting Started
When the program PHREEQC is invoked, two files are used to define the thermodynamic model and the types of calculations that will be done, the database file and the input file. The database file is read once (to the end of the file or until an
END
keyword is encountered) at the beginning of the program. The input file is then read and processed simulation by simulation (as defined by
END
keywords) until the end of the file. The formats for the keyword data blocks are the same for either the input file or the database file.
The database file is used to define static data for the thermodynamic model. Although any keyword data block can occur in the database file, normally, the file contains the keyword data blocks:
EXCHANGE_MASTER_SPECIES
,
EXCHANGE_SPECIES
,
PHASES
,
RATES
,
SOLUTION_MASTER_SPECIES
,
SOLUTION_SPECIES
,
SURFACE_MASTER_SPECIES
, and
SURFACE_SPECIES
. These keyword data blocks define rate expressions, master species, and the stoichiometric and thermodynamic properties of all of the aqueous phase species, exchange species, surface species, and pure phases.
Nine database files are provided with the program: (1) phreeqc.dat, a database file derived from PHREEQE (Parkhurst and others, 1980), which is consistent with wateq4f.dat, but has a smaller set of elements and aqueous species (table 3); (2) Amm.dat is the same as phreeqc.dat, except that ammonia redox state has been decoupled from the rest of the nitrogen system; that is, ammonia has been defined as a separate component; (3) wateq4f.dat, a database file derived from WATEQ4F (Ball and Nordstrom, 1991); (4) llnl.dat, a database file derived from databases for EQ3/6 and Geochemist’s Workbench that uses thermodynamic data compiled by the Lawrence Livermore National Laboratory; (5) minteq.dat, a database derived from the databases for the program MINTEQA2 (Allison and others, 1990); (6) minteq.v4.dat, a database derived from MINTEQA2 version 4 (U.S. Environmental Protection Agency, 1998); (7) pitzer.dat, a database for the specific-ion-interaction model of Pitzer (Pitzer, 1973) as implemented in PHRQPITZ (Plummer and others, 1988); (8) sit.dat, a database implementing the Specific ion Interaction Theory (SIT) as described by Grenthe and others (1997); and (9) iso.dat, a partial implementation of the individual component approach to isotope calculations as described by Thorstenson and Parkhurst (2002, 2004). The elements and element valence states, corresponding notation, and default formula used to convert mass concentration to mole concentration units in the database phreeqc.dat are listed in table 3. Other databases may use different sets of elements, different notation for the element names, or different default conversion formulas.
Table 3. Elements and element valence states included in default database
phreeqc.dat
, including PHREEQC notation and default formula for gram formula weight.
[For alkalinity, formula for gram equivalent weight is given]
|
Element or element valence state
|
PHREEQC
notation
|
Formula used for default gram formula weight
|
|
Alkalinity
|
Alkalinity
|
Ca
0.5
(CO
3
)
0.5
|
|
Aluminum
|
Al
|
Al
|
|
Barium
|
Ba
|
Ba
|
|
Boron
|
B
|
B
|
|
Bromide
|
Br
|
Br
|
|
Cadmium
|
Cd
|
Cd
|
|
Calcium
|
Ca
|
Ca
|
|
Carbon
|
C
|
HCO
3
|
|
Carbon(IV)
|
C(4)
|
HCO
3
|
|
Carbon(-IV), methane
|
C(-4)
|
CH
4
|
|
Chloride
|
Cl
|
Cl
|
|
Copper
|
Cu
|
Cu
|
|
Copper(II)
|
Cu(2)
|
Cu
|
|
Copper(I)
|
Cu(1)
|
Cu
|
|
Fluoride
|
F
|
F
|
|
Hydrogen(0), dissolved hydrogen
|
H(0)
|
H
|
|
Iron
|
Fe
|
Fe
|
|
Iron(II)
|
Fe(2)
|
Fe
|
|
Iron(III)
|
Fe(3)
|
Fe
|
|
Lead
|
Pb
|
Pb
|
|
Lithium
|
Li
|
Li
|
|
Magnesium
|
Mg
|
Mg
|
|
Manganese
|
Mn
|
Mn
|
|
Manganese(II)
|
Mn(2)
|
Mn
|
|
Manganese(III)
|
Mn(3)
|
Mn
|
|
Nitrogen
|
N
|
N
|
|
Nitrogen(V), nitrate
|
N(5)
|
N
|
|
Nitrogen(III), nitrite
|
N(3)
|
N
|
|
Nitrogen(0), dissolved nitrogen
|
N(0)
|
N
|
|
Nitrogen(-III), ammonia
|
N(-3)
|
N
|
|
Oxygen(0), dissolved oxygen
|
O(0)
|
O
|
|
Phosphorus
|
P
|
P
|
|
Potassium
|
K
|
K
|
|
Silica
|
Si
|
SiO
2
|
|
Sodium
|
Na
|
Na
|
|
Strontium
|
Sr
|
Sr
|
|
Sulfur
|
S
|
SO
4
|
|
Sulfur(VI), sulfate
|
S(6)
|
SO
4
|
|
Sulfur(-II), sulfide
|
S(-2)
|
S
|
|
Zinc
|
Zn
|
Zn
|
The input data file is used (1) to define the types of calculations that are to be done, and (2) if necessary, to modify the data read from the database file. If new elements and aqueous species, exchange species, surface species, or phases need to be included in addition to those defined in the database file, or if the stoichiometry, log
K
, or activity coefficient information from the database file needs to be modified for a given run, then the keywords mentioned in the previous paragraph can be included in the input file. The data read for these keyword data blocks in the input file will augment or supersede the data read from the database file. In many cases, the thermodynamic model defined in the database will not be modified, and the above keywords will not be used in the input data file.
The place to start is with the simplest input file, which contains only a
SOLUTION
data block containing the dissolved concentrations of elements. With this input file, PHREEQC will perform a speciation calculation and calculate saturation indices for the solution. More complex calculations will calculate new solution compositions as a function of reactions. Reactions can be understood as occurring in a beaker, where a solution (as defined by a
SOLUTION
data block) is placed in the beaker, and then additional reactants are added. The reactants are defined with the keywords
EQUILIBRIUM_PHASES
,
EXCHANGE
,
GAS_PHASE
,
KINETICS
,
REACTION
,
SOLID_SOLUTIONS
, and
SURFACE
. One or more of these reactants may be added to the beaker, and then system equilibrium is calculated, which results in mole transfers into and out of solution, and new pH and element concentrations. The pressure and temperature of the reaction may be defined with
REACTION_PRESSURE
and
REACTION_TEMPERATURE
. So, the design of PHREEQC is fairly intuitive. You must choose the composition of a starting solution and then decide which types of reactants you need to add to the beaker to model your system. Transport reactions are simply defined by a series of beakers, each containing a set of reactants, and water flows and mixes from one beaker to the next and equilibrates with the reactants in each beaker in sequence.
Units
The concentrations of elements in solution and the mass of water in the solution are defined through the
SOLUTION
or SOLUTION_SPREAD data block. Internally, all concentrations are converted to molality and the number of moles of each element in solution (including hydrogen and oxygen) is calculated from the molalities and the mass of water. Thus, internally, a solution is simply a list of elements and the number of moles of each element.
PHREEQC allows each reactant to be defined independently. In particular, reactants (
EQUILIBRIUM_PHASES
,
EXCHANGE
,
GAS_PHASE
,
KINETICS
,
REACTION
, SOLID_SOLUTIONS, and
SURFACE
) are defined in terms of moles, without reference to a volume or mass of water. Systems are defined by combining a solution with a set of reactants that react either reversibly (
EQUILIBRIUM_PHASES
,
EXCHANGE
,
GAS_PHASE
, SOLID_SOLUTIONS, and
SURFACE
) or irreversibly (
KINETICS
or
REACTION
). Essentially, all of the moles of elements in the solution and the reversible reactants are combined, the moles of irreversible reactants are added (or removed), and a new system equilibrium is calculated. Only after system equilibrium is calculated is the mass of water in the system known, and only then the molalities of all entities can be calculated.
For transport calculations, each cell is a system that is defined by the solution and all the reactants contained in keywords that bear the same number as the cell number. The system for the cell initially is defined by the moles of elements that are present in the solution and the moles of each reactant. The compositions of all these entities evolve as the transport calculations proceed.
Keywords
The following sections describe the data input requirements for the program. Each type of data is input through a specific keyword data block. Most keywords are listed in alphabetical order within this section of the report; however, a set of keywords most pertinent to model developers is described in See Appendix A. Keyword Data Blocks for Programmers. Each keyword data block may have a number of identifiers, many of which are optional. Identifiers may be entered in any order; the line numbers given in examples for the keyword data blocks are for identification purposes only. Default values for identifiers are used if the identifier is omitted.
| Next || Previous || Top |